Figure 2a#
LARRY Dataset#
Preprocessing and dimension reduction of the full dataset
[1]:
%load_ext nb_black
import scdiffeq as sdq
h5ad_path = "../KleinLabData/in_vitro/adata.Weinreb2020.in_vitro.gene_filtered.h5ad"
adata = sdq.io.read_h5ad(h5ad_path)
AnnData object with n_obs × n_vars = 130887 × 25289
obs: 'Library', 'Cell barcode', 'Time point', 'Starting population', 'Cell type annotation', 'Well', 'SPRING-x', 'SPRING-y', 'clone_idx', 'fate_observed', 't0_fated'
var: 'gene_ids', 'hv_genes', 'use_genes'
uns: 'fate_counts', 'time_occupance'
obsm: 'X_clone', 'cell_fate_df'
[2]:
adata = adata[:, adata.var["use_genes"]].copy()
adata
[2]:
AnnData object with n_obs × n_vars = 130887 × 2447
obs: 'Library', 'Cell barcode', 'Time point', 'Starting population', 'Cell type annotation', 'Well', 'SPRING-x', 'SPRING-y', 'clone_idx', 'fate_observed', 't0_fated'
var: 'gene_ids', 'hv_genes', 'use_genes'
uns: 'fate_counts', 'time_occupance'
obsm: 'X_clone', 'cell_fate_df'
[3]:
reducer = sdq.tl.DimensionReduction(
adata, save_path="./LARRY_full_dataset.reducer_models"
)
- [INFO] | mkdir: ./LARRY_full_dataset.reducer_models
[4]:
reducer.fit_scaler()
- [INFO] | Configuring: SCALER model
- [INFO] | PATH: ./LARRY_full_dataset.reducer_models/scaler_model.pkl DOES NOT EXIST.
- [INFO] | Fitting SCALER model
- [INFO] | Saving SCALER model to: ./LARRY_full_dataset.reducer_models/scaler_model.pkl
[5]:
reducer.fit_pca()
- [INFO] | Configuring: PCA model
- [INFO] | PATH: ./LARRY_full_dataset.reducer_models/pca_model.pkl DOES NOT EXIST.
- [INFO] | Fitting PCA model
- [INFO] | Saving PCA model to: ./LARRY_full_dataset.reducer_models/pca_model.pkl
[6]:
reducer.fit_umap()
- [INFO] | Configuring: UMAP model
- [INFO] | PATH: ./LARRY_full_dataset.reducer_models/umap_model.pkl DOES NOT EXIST.
- [INFO] | Fitting UMAP model
- [INFO] | Saving UMAP model to: ./LARRY_full_dataset.reducer_models/umap_model.pkl
[7]:
adata
[7]:
AnnData object with n_obs × n_vars = 130887 × 2447
obs: 'Library', 'Cell barcode', 'Time point', 'Starting population', 'Cell type annotation', 'Well', 'SPRING-x', 'SPRING-y', 'clone_idx', 'fate_observed', 't0_fated', 'train'
var: 'gene_ids', 'hv_genes', 'use_genes'
uns: 'fate_counts', 'time_occupance'
obsm: 'X_clone', 'cell_fate_df', 'X_pca', 'X_umap'
layers: 'X_scaled'
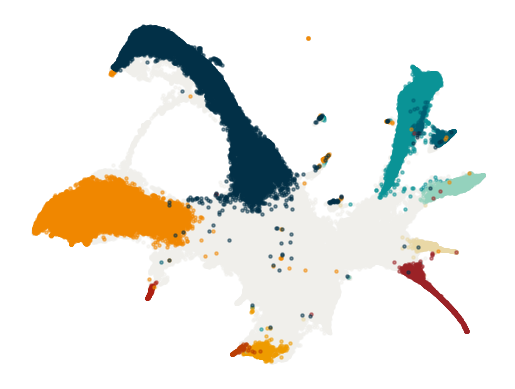
Plot#
[10]:
import matplotlib.pyplot as plt
import scdiffeq_plots as sdq_pl
import larry
cmap = larry.pl.in_vitro_cmap()
df = adata.obs.copy()
[23]:
fig, axes = sdq_pl.plot(delete_spines=["all"], rm_ticks=True)
for en, (group, group_df) in enumerate(df.groupby("Cell type annotation")):
X_umap = adata[group_df.index].obsm["X_umap"]
c = cmap.loc[cmap["Cell type annotation"] == group]["color"]
if group in ["undiff", "Undifferentiated"]:
z = 0
else:
z = en + 2
axes[0].scatter(X_umap[:, 0], X_umap[:, 1], s=5, alpha=0.5, c=c, zorder=z)