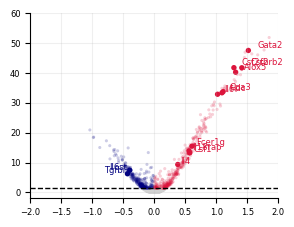
Figure 3E, F, G#
Import packages#
[1]:
%load_ext nb_black
import scdiffeq as sdq
import cellplots as cp
import matplotlib.pyplot as plt
import matplotlib.cm as cm
import pandas as pd
import numpy as np
import cell_perturb
import scipy.stats
import sklearn
import larry
import pathlib
import glob
import torch
import matplotlib.cm as cm
import statsmodels.stats.meta_analysis
from scipy.stats import combine_pvalues
import seaborn as sns
from typing import Union, Dict, List
cmap = larry.pl.InVitroColorMap()._dict
h5ad_path = (
"/home/mvinyard/data/adata.reprocessed_19OCT2023.more_feature_inclusive.h5ad"
)
adata = sdq.io.read_h5ad(h5ad_path)
AnnData object with n_obs × n_vars = 130887 × 2492
obs: 'Library', 'Cell barcode', 'Time point', 'Starting population', 'Cell type annotation', 'Well', 'SPRING-x', 'SPRING-y', 'clone_idx', 'fate_observed', 't0_fated', 'train'
var: 'gene_ids', 'hv_gene', 'must_include', 'exclude', 'use_genes'
uns: 'fate_counts', 'h5ad_path', 'time_occupance'
obsm: 'X_clone', 'X_pca', 'X_umap', 'cell_fate_df'
layers: 'X_scaled'
[2]:
def aggr_fate_statistics(results, fate):
_aggr_fate_frames = []
for version, frame in results.items():
fate_version_frame = (
frame["lfc"][fate]
.to_frame()
.reset_index()
.rename({"index": "gene", fate: "lfc"}, axis=1)
)
fate_version_frame["model"] = version
fate_version_frame["pval"] = frame["pval"][fate].values
fate_version_frame["se"] = (
frame["lfc_std"][fate].div(np.sqrt(frame["lfc_std"]["n"])).values
)
_aggr_fate_frames.append(fate_version_frame)
return pd.concat(_aggr_fate_frames)
def adjust_negative_i2(meta_results_df):
i2 = np.zeros(len(meta_results_df))
mask = meta_results_df["i2"] > 0
pos_i2 = meta_results_df.loc[mask]["i2"].values
i2[mask] = pos_i2
meta_results_df["i2"] = i2
def run_meta_analysis(aggr_stats):
meta_analysis_results = []
for gene in aggr_stats["gene"].unique():
gene_df = aggr_stats.loc[aggr_stats["gene"] == gene]
effect = gene_df["lfc"].values
variance = (gene_df["se"] ** 2).values
res_re = statsmodels.stats.meta_analysis.combine_effects(
effect=effect, variance=variance, method_re="iterated"
)
conf_int = np.array(res_re.conf_int())
ci_low, ci_high = conf_int[:, 0], conf_int[:, 1]
meta_analysis_results.append(
{
"gene": gene,
"pooled_mean_lfc_re": res_re.mean_effect_re,
"ci_low": ci_low[0], # Accessing the first element for CI
"ci_high": ci_high[0], # Accessing the first element for CI
"i2": res_re.i2,
"q": res_re.q,
}
)
meta_results_df = pd.DataFrame(meta_analysis_results).set_index("gene")
adjust_negative_i2(meta_results_df)
return meta_results_df
def combine_expand(pvals):
stat, combined_pval = combine_pvalues(pvals, method="fisher")
return {"stat": stat, "combined_pval": combined_pval}
def prepare_stat_df_for_plotting(stats):
stats["x"] = stats["pooled_mean_lfc_re"]
stats["y"] = stats["combined_pval"].apply(np.log10) * -1
stats["significant"] = stats["combined_pval"] < 0.05
stats["insignificant"] = stats["combined_pval"] >= 0.05
stats["pos"] = stats["pooled_mean_lfc_re"] > 0
stats["neg"] = stats["pooled_mean_lfc_re"] <= 0
stats["sig.pos"] = stats["significant"] & stats["pos"]
stats["sig.neg"] = stats["significant"] & stats["neg"]
stats["insig.pos"] = stats["insignificant"] & stats["pos"]
stats["insig.neg"] = stats["insignificant"] & stats["neg"]
assert not any(stats[["sig.pos", "sig.neg", "insig.pos", "insig.neg"]].sum(1) > 1)
stats["group"] = stats[["sig.pos", "sig.neg", "insig.pos", "insig.neg"]].idxmax(1)
return stats
def plot(stats, xlim=(None, None), ylim=(-2, None)):
fig, axes = cp.plot(
1,
1,
height=0.5,
width=0.5,
delete=[["top", "right"]],
title=[
""
], # LARRY Neutrophil Fate Perturbation Screen (N=5, z=10)"], # Meta (5 models):
)
for group, group_df in stats.groupby("group"):
if "insig" in group:
c = "lightgrey"
elif "pos" in group:
c = "crimson"
elif "neg" in group:
c = "navy"
axes[0].scatter(
group_df["x"],
group_df["y"],
s=5,
ec="None",
c=c,
rasterized=True,
alpha=0.2,
)
axes[0].grid(True, alpha=0.2)
axes[0].set_xlim(xlim)
axes[0].set_ylim(ylim)
neg_highlight = stats[stats["sig.neg"]].sort_values("y").tail(15)
for i, row in neg_highlight.iterrows():
axes[0].scatter(row["x"], row["y"], s=15, c="navy", ec="None", rasterized=True)
axes[0].text(
x=row["x"] * 1.1,
y=row["y"] * 1.02,
s=row.name,
color="navy",
ha="right",
fontsize=6,
)
neg_highlight = stats[stats["sig.pos"]].sort_values("y").tail(15)
for i, row in neg_highlight.iterrows():
axes[0].scatter(
row["x"], row["y"], s=15, c="crimson", ec="None", rasterized=True
)
axes[0].text(
x=row["x"] * 1.1, y=row["y"] * 1.02, s=row.name, color="crimson", fontsize=6
)
axes[0].hlines(y=-np.log10(0.05), xmin=-2, xmax=2, color="k", ls="--", lw=1)
# plt.savefig("LARRY.neu_screen.meta_analysis.svg", dpi=500)
[3]:
import ABCParse
class RawScreenProcessor(ABCParse.ABCParse):
"""Processed pkl to .csv"""
def __init__(self, results_dir: Union[pathlib.Path, str], *args, **kwargs):
self.__parse__(locals())
self._initialize_directory_structure()
@property
def results_dir(self):
return pathlib.Path(self._results_dir)
@property
def pkl_dir(self):
return self.results_dir.joinpath("pkl")
@property
def csv_dir(self):
return self.results_dir.joinpath("csv")
def _initialize_directory_structure(self):
"""/pkl/ subdir should already exist, so we shouldn't create it here."""
if not self.csv_dir.exists():
self.csv_dir.mkdir()
@property
def _discovered_csv_paths(self):
return sorted(list(self.csv_dir.glob("*.csv")))
@property
def pkl_paths(self):
return sorted(list(self.pkl_dir.glob("*.pkl")))
def _process_attribute(self, file, key):
return (
pd.DataFrame({gene: result.stats[key] for gene, result in file.items()})
.fillna(0)
.T
)
def _get_n(self, file, gene):
return np.unique([file[gene].ctrl.shape[1], file[gene].prtb.shape[1]])[0]
def _read_and_process_raw_pkl_files(self):
ScreenResults = {}
attr_cols = ["lfc", "lfc_std", "pval"]
for pkl_path in self.pkl_paths:
version = pkl_path.name.split("version_")[-1].split(".")[0]
version = f"version_{version}"
if not version in ScreenResults:
ScreenResults[version] = {}
csv_paths = {
attr: self.csv_dir.joinpath(f"{version}.{attr}.csv")
for attr in attr_cols
}
exists = [csv_path.exists() for csv_path in list(csv_paths.values())]
if not all(exists):
file = sdq.io.read_pickle(pkl_path)
for attr, csv_path in csv_paths.items():
df = self._process_attribute(file, attr)
if attr == "lfc_std":
n = [self._get_n(file, gene) for gene in file.keys()]
df["n"] = n
df.to_csv(csv_path)
ScreenResults[version][attr] = df
else:
for attr, csv_path in csv_paths.items():
ScreenResults[version][attr] = pd.read_csv(csv_path, index_col=0)
return ScreenResults
def __call__(self, *args, **kwargs):
self.__update__(locals())
return self._read_and_process_raw_pkl_files()
def process_raw_results(results_dir: str = "./larry_perturbation_screen_results/"):
raw_screen_processor = RawScreenProcessor(results_dir=results_dir)
return raw_screen_processor()
[4]:
def aggr_fate_statistics(results, fate):
_aggr_fate_frames = []
for version, frame in results.items():
fate_version_frame = (
frame["lfc"][fate]
.to_frame()
.reset_index()
.rename({"index": "gene", fate: "lfc"}, axis=1)
)
fate_version_frame["model"] = version
fate_version_frame["pval"] = frame["pval"][fate].values
fate_version_frame["se"] = (
frame["lfc_std"][fate].div(np.sqrt(frame["lfc_std"]["n"])).values
)
_aggr_fate_frames.append(fate_version_frame)
return pd.concat(_aggr_fate_frames)
def adjust_negative_i2(meta_results_df):
i2 = np.zeros(len(meta_results_df))
mask = meta_results_df["i2"] > 0
pos_i2 = meta_results_df.loc[mask]["i2"].values
i2[mask] = pos_i2
meta_results_df["i2"] = i2
def run_meta_analysis(aggr_stats):
meta_analysis_results = []
for gene in aggr_stats["gene"].unique():
gene_df = aggr_stats.loc[aggr_stats["gene"] == gene]
effect = gene_df["lfc"].values
variance = (gene_df["se"] ** 2).values
res_re = statsmodels.stats.meta_analysis.combine_effects(
effect=effect, variance=variance, method_re="iterated"
)
conf_int = np.array(res_re.conf_int())
ci_low, ci_high = conf_int[:, 0], conf_int[:, 1]
meta_analysis_results.append(
{
"gene": gene,
"pooled_mean_lfc_re": res_re.mean_effect_re,
"ci_low": ci_low[0], # Accessing the first element for CI
"ci_high": ci_high[0], # Accessing the first element for CI
"i2": res_re.i2,
"q": res_re.q,
}
)
meta_results_df = pd.DataFrame(meta_analysis_results).set_index("gene")
adjust_negative_i2(meta_results_df)
return meta_results_df
def combine_expand(pvals):
stat, combined_pval = combine_pvalues(pvals, method="fisher")
return {"stat": stat, "combined_pval": combined_pval}
def prepare_stat_df_for_plotting(stats):
stats["x"] = stats["pooled_mean_lfc_re"]
stats["y"] = stats["combined_pval"].apply(np.log10) * -1
stats["significant"] = stats["combined_pval"] < 0.05
stats["insignificant"] = stats["combined_pval"] >= 0.05
stats["pos"] = stats["pooled_mean_lfc_re"] > 0
stats["neg"] = stats["pooled_mean_lfc_re"] <= 0
stats["sig.pos"] = stats["significant"] & stats["pos"]
stats["sig.neg"] = stats["significant"] & stats["neg"]
stats["insig.pos"] = stats["insignificant"] & stats["pos"]
stats["insig.neg"] = stats["insignificant"] & stats["neg"]
assert not any(stats[["sig.pos", "sig.neg", "insig.pos", "insig.neg"]].sum(1) > 1)
stats["group"] = stats[["sig.pos", "sig.neg", "insig.pos", "insig.neg"]].idxmax(1)
return stats
def plot_volcano(
stats, xlim=(None, None), ylim=(-2, None), savepath=None, highlight_genes=[]
):
fig, axes = cp.plot(
1,
1,
height=0.5,
width=0.5,
delete=[["top", "right"]],
title=[
""
], # LARRY Neutrophil Fate Perturbation Screen (N=5, z=10)"], # Meta (5 models):
)
for group, group_df in stats.groupby("group"):
if "insig" in group:
c = "lightgrey"
elif "pos" in group:
c = "crimson"
elif "neg" in group:
c = "navy"
axes[0].scatter(
group_df["x"],
group_df["y"],
s=5,
ec="None",
c=c,
rasterized=True,
alpha=0.2,
)
axes[0].grid(True, alpha=0.2)
axes[0].set_xlim(xlim)
axes[0].set_ylim(ylim)
stats_highlight = stats.loc[highlight_genes].copy()
neg_highlight = stats_highlight[stats_highlight["sig.neg"]]
for i, row in neg_highlight.iterrows():
axes[0].scatter(row["x"], row["y"], s=15, c="navy", ec="None", rasterized=True)
axes[0].text(
x=row["x"] * 1.1,
y=row["y"] * 1.02,
s=row.name,
color="navy",
ha="right",
fontsize=6,
)
pos_highlight = stats_highlight[stats_highlight["sig.pos"]]
for i, row in pos_highlight.iterrows():
axes[0].scatter(
row["x"], row["y"], s=15, c="crimson", ec="None", rasterized=True
)
axes[0].text(
x=row["x"] * 1.1, y=row["y"] * 1.02, s=row.name, color="crimson", fontsize=6
)
axes[0].hlines(y=-np.log10(0.05), xmin=-2, xmax=2, color="k", ls="--", lw=1)
if savepath:
plt.savefig(savepath, dpi=500)
[5]:
import ABCParse
class MetaAnalysis(ABCParse.ABCParse):
def __init__(
self, results_dir: str = "./larry_perturbation_screen_results", *args, **kwargs
):
self.__parse__(locals())
self.results = process_raw_results(self.results_dir)
@property
def results_dir(self):
results_dir = pathlib.Path(self._results_dir)
if not results_dir.exists():
results_dir.mkdir()
return results_dir
def _combine_pvals(self, aggr):
return aggr.groupby("gene")["pval"].apply(combine_expand).unstack()
@property
def aggr(self):
if not hasattr(self, "_attr"):
self._attr = aggr_fate_statistics(self.results, fate=self._fate)
return self._attr
def forward(self, fate):
self._fate = fate
meta = run_meta_analysis(self.aggr)
combined_pvals = self._combine_pvals(self.aggr)
stats = pd.concat(
[
meta["pooled_mean_lfc_re"],
combined_pvals["combined_pval"],
],
axis=1,
)
return prepare_stat_df_for_plotting(stats)
[6]:
neu_meta_analysis = MetaAnalysis()
neu_stats = neu_meta_analysis.forward("Neutrophil")
neu_aggr = neu_meta_analysis.aggr
[7]:
mon_meta_analysis = MetaAnalysis()
mon_stats = mon_meta_analysis.forward("Monocyte")
mon_aggr = mon_meta_analysis.aggr
[8]:
baso_meta_analysis = MetaAnalysis()
baso_stats = baso_meta_analysis.forward("Baso")
baso_aggr = baso_meta_analysis.aggr
[9]:
HighlightGenes = {
"Neutrophil": [
"Il6st",
"Socs3",
"Ccr2",
"Elane",
"Mpo",
"Prtn3",
"Ctsg",
"Gfi1",
"S100a8",
"S100a9",
"Cd33",
"Itgam",
"Cebpe",
"Lipg",
"Tfrc",
"Gpx4",
"Cd47",
"Lcn2",
"Itgb2",
"Ncf1",
"Cebpd",
"Mcl1",
],
"Monocyte": [
"Csf1r",
"Ccl3",
"Klf4",
"Socs3",
"Vim",
"Ccr2",
"Gata2",
"Il6st",
"Cebpa",
"Tgfbi",
"Il6",
"Fpr1",
"Gfi1",
"Cxcr2",
"Cd47",
"Igf1r",
"Hgf",
"Lpl",
],
"Baso": [
"Csf2rb",
"Csf2rb2",
"Il4",
"Csf1",
"Il6",
"Gata2",
"Cpa3",
"Fcer1g",
"Hdc",
"Il18rap",
"Il6st",
"Tgfbi",
"Alox5",
],
}
[10]:
plot_volcano(
neu_stats,
xlim=(-1.5, 1.5),
ylim=(-2, 60),
highlight_genes=HighlightGenes["Neutrophil"],
savepath="LARRY.neu_screen.meta_analysis.svg",
)
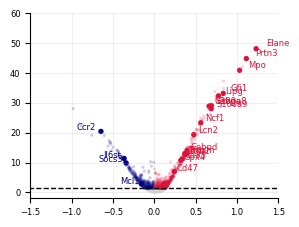
[12]:
plot_volcano(
mon_stats,
xlim=(-1.5, 1.5),
ylim=(-2, 60),
highlight_genes=HighlightGenes["Monocyte"],
savepath="LARRY.mon_screen.meta_analysis.svg",
)
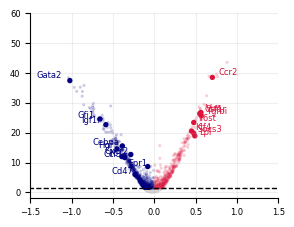
[13]:
plot_volcano(
baso_stats,
xlim=(-2, 2),
ylim=(-2, 60),
highlight_genes=HighlightGenes["Baso"],
savepath="LARRY.baso_screen.meta_analysis.svg",
)