Figure 4G#
Import packages#
[1]:
import anndata
import ezplot
import matplotlib.pyplot as plt
import pandas as pd
import glob
import pathlib
import seaborn as sns
import sklearn.cluster
import numpy as np
import matplotlib.cm as cm
Define helper functions#
[3]:
def read_gene_velocity_corr(idx: int, version: int):
base_path = f"./version_{version}_gene_velocity_corr/{idx}.*.csv"
paths = glob.glob(base_path)
DataFrames = {}
for path in paths:
fpath = pathlib.Path(path)
fate = fpath.name.split(".")[1]
if fate != "Undifferentiated":
DataFrames[fate] = pd.read_csv(path, index_col = 0)
return DataFrames
def _adjust_colnames(data, fate: str = "Monocyte"):
df = data[fate].copy()
df.columns = [f"{fate}.{key}" for key in df.columns]
return df
Two fates#
For the two-fate example, we’ll use idx=19977 (v3)
[4]:
data = read_gene_velocity_corr(idx = 19977, version = 3)
df = pd.concat([_adjust_colnames(data, "Monocyte"), _adjust_colnames(data, "Neutrophil")], axis = 1)
df
[4]:
| Monocyte.f.corr | Monocyte.f.pval | Monocyte.g.corr | Monocyte.g.pval | Neutrophil.f.corr | Neutrophil.f.pval | Neutrophil.g.corr | Neutrophil.g.pval | |
|---|---|---|---|---|---|---|---|---|
| gene_ids | ||||||||
| 1110002J07Rik | 0.246731 | 0.119912 | 0.594733 | 4.121254e-05 | 0.025470 | 8.744020e-01 | 0.406625 | 8.333975e-03 |
| 1110032F04Rik | 0.537274 | 0.000292 | 0.322823 | 3.952964e-02 | 0.533234 | 3.310762e-04 | 0.298061 | 5.838780e-02 |
| 1500002F19Rik | 0.199835 | 0.210322 | -0.192597 | 2.276558e-01 | -0.679190 | 1.050207e-06 | -0.932045 | 8.675594e-19 |
| 1500026H17Rik | -0.144147 | 0.368571 | -0.669251 | 1.718679e-06 | -0.656420 | 3.160551e-06 | -0.927592 | 2.872365e-18 |
| 1600010M07Rik | -0.666030 | 0.000002 | -0.851748 | 1.666314e-12 | -0.297194 | 5.915940e-02 | -0.664837 | 2.126375e-06 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... |
| Zmpste24 | 0.667209 | 0.000002 | 0.913492 | 8.110006e-17 | -0.876496 | 5.967734e-14 | -0.819266 | 5.826618e-11 |
| Zmynd15 | -0.038569 | 0.810783 | 0.464953 | 2.192460e-03 | 0.329221 | 3.556725e-02 | 0.801130 | 3.157972e-10 |
| Znfx1 | -0.624361 | 0.000013 | -0.976683 | 1.131776e-27 | -0.771510 | 3.547780e-09 | -0.961632 | 1.633703e-23 |
| Zscan2 | 0.462753 | 0.002316 | 0.499486 | 8.850821e-04 | 0.349633 | 2.504204e-02 | 0.112174 | 4.850126e-01 |
| Zyx | -0.538328 | 0.000283 | -0.969048 | 2.649156e-25 | -0.642644 | 5.891798e-06 | -0.944852 | 1.659873e-20 |
2492 rows × 8 columns
Read table of transcription factors (from Weinreb2020)#
[5]:
def get_tf_table(df):
tf_df = pd.read_table(
"/Users/michaelvinyard/tf_list.txt",
usecols=[1, 2, 3],
index_col = 0,
header=None,
names=['idx', 'gene_name', 'description'],
)
return df.loc[df.index.isin(tf_df['gene_name'].tolist())]
[6]:
tf_df = get_tf_table(df)
tf_df = tf_df.filter(regex = "corr").copy()
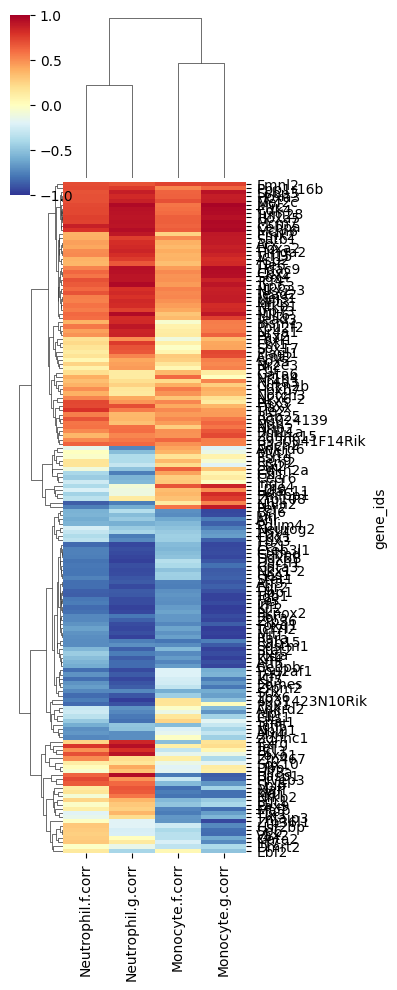
Plot heatmap for Figure 4G#
[7]:
cg = sns.clustermap(
tf_df,
vmin = -1,
vmax = 1,
cmap = cm.RdYlBu_r,
figsize = (4, 10),
rasterized = True,
yticklabels = tf_df.index,
)
plt.savefig("Figure4G.svg")
[8]:
tf_df.to_csv("scdiffeq.SupplementaryTable6.csv")
[ ]: